
A Peptide Case Study. What Semax Reveals About the U.S. Drug Pipeline
The U.S. drug pipeline is structurally blind to certain categories of evidence.


Peptides have become increasingly popular among longevity and wellness enthusiasts. The preclinical data on many of these compounds are substantial, as are anecdotal reports of benefit. However, most have not undergone clinical testing. In 2023, the FDA placed these products in its high-risk Category 2, blocking compounding pharmacies from producing them. A grey market developed to fill the gap, with hundreds of thousands to potentially low millions of Americans ordering research chemicals for personal medical use. In April, the FDA removed twelve from the list, and an advisory committee will soon review whether they belong on the list available to compounding pharmacies.
Compounded products will be higher quality than what the grey market offers. The harder question is upstream: if these compounds work, why have they never been approved? One of the twelve is Semax, a Russian-developed peptide with a thirty-year clinical record for ischemic stroke. The deeper question is not on the agenda: why a molecule with three decades of foreign clinical use has no path into American medicine. The prior piece in this series covered the full list and the regulatory mechanics. Semax is the case study for how the system fails to evaluate compounds like these.
If you enjoy these posts, consider subscribing and becoming a part of our growing community!
A man in his sixties is wheeled into a stroke unit at a Moscow hospital with weakness on his right side and slurred speech. The neurologist on call follows the standard acute-ischemic protocol: imaging, tPA if the window allows, mechanical thrombectomy if indicated, and a course of intranasal Semax administered alongside the rest. Down the hall, the pharmacist does not blink. The drug has been on the Russian List of Vital and Essential Medicines since 2011. It has been used for stroke in Russia since the late 1990s.
Same week, an academic medical center in Cleveland. A man in his sixties presents with weakness on his right side and slurred speech. The neurologist runs the U.S. acute-ischemic protocol: imaging, tPA if the window allows, mechanical thrombectomy if a large-vessel occlusion is confirmed. Semax does not enter the conversation. It cannot. The molecule is not approved by the FDA, has no U.S. sponsor, and exists in American medicine somewhere between an unscheduled research chemical and a gray-market peptide sold to biohackers on the internet.
Same drug. Same indication. Three decades of post-marketing exposure in one country and a regulatory dead silence in the other.
In April 2026, the FDA moved Semax, along with eleven other peptides, off Category 2 of its Section 503A bulks list. The Pharmacy Compounding Advisory Committee will hear the Semax petition in July. That is news. But it is only part of the story. It does not approve Semax for stroke or for any other indication. It clears the molecule for a public review of whether it can be compounded by pharmacies, a question two regulatory floors down from “does it work.”
There are real questions. The original Semax trial base was thinner than its proponents claim, and the methodological standards behind it would likely not satisfy a modern FDA reviewer. The molecule may or may not work. That is not the question.
The question is why the U.S. system has no way to even ask.
The compound
Semax was designed in 1982 at the Institute of Molecular Genetics, then part of the Soviet Academy of Sciences. It was first described in the open scientific literature in 1991 (Potaman et al., 1991). Its structure is a synthetic heptapeptide, Met-Glu-His-Phe-Pro-Gly-Pro, built around the cognitive-active core of adrenocorticotropic hormone, residues 4 through 7, with a Pro-Gly-Pro extension that resists peptidase cleavage. The modification extends the half-life from minutes to hours, eliminates the steroidogenic activity of the parent hormone, and produces measurable CNS effects when delivered intranasally.
The initial Phase I trials ran from 1990 to 1994, followed by Phase II through 1996. Russian Ministry of Health registration came in the mid-to-late 1990s. The on-label indications include acute ischemic stroke, transient ischemic attack, optic nerve atrophy, and a cluster of cognitive and cerebrovascular conditions. The mechanism is officially listed as not fully characterized. The published work converges on two candidate effects: induction of BDNF and NGF expression with concurrent upregulation of the TrkB receptor, and modulation of dopaminergic and serotonergic signaling. Russia added Semax to its List of Vital and Essential Drugs on December 7, 2011.
The molecule has accumulated decades of broad clinical population exposure in Russian post-marketing surveillance, with more cumulative patient-years than many compounds the FDA has approved. It is not a regulatory orphan in the United States because it has been ignored. It is a regulatory orphan because the U.S. system has no machinery for evaluating it.
The thesis
The U.S. drug development pipeline is a sequence of filters. Each one determines which compounds and which evidence reach the system. Most are individually defensible. None are individually about whether a drug works.
Semax fails seven of these filters simultaneously. That pattern, not the molecule itself, is the story.
No sponsor. Drugs do not get approved. Sponsors get drugs approved. Every FDA-approved compound has a corporate or institutional entity behind it that funds the trials, files the IND, runs the regulatory correspondence, manages the post-marketing commitments, and absorbs the cost of failure. Semax has none of this. There is no U.S. licensing biotech. There is no academic champion preparing an investigator-led IND. There is no patient advocacy foundation pushing the indication. There is no sell-side analyst tracking the program. The infrastructure of people who normally drag a foreign drug through approval does not exist for this molecule.
The ICH boundary. The FDA accepts foreign clinical data under specific conditions tied to compliance with International Council for Harmonisation guidelines, GCP-compliant auditing, and population comparability. Russia is not an ICH member. The bulk of the Semax evidence base sits in Russian-language journals, generated under late-Soviet and post-Soviet trial standards, that the agency has no established framework for evaluating without commissioning independent verification. No one is paying for that verification. No one has reason to.
Sanctions and the post-2022 IP collapse. Until 2022, a U.S. biotech could plausibly have licensed Semax from the Russian Academy of Sciences and run a Western development program. That pathway is now closed. Sanctions have made technology transfer from state-affiliated Russian institutions legally and politically difficult. Pharmaceutical investors will not write checks against deals with Russian counterparties. The IP-level transaction that would need to happen first is not currently doable, and there is no obvious horizon on which it becomes doable again.
The neuroprotectant graveyard. The recent history of acute-stroke neuroprotection is a wreckage. NXY-059 failed Phase 3 in the SAINT-II trial in the New England Journal of Medicine in 2007 after positive earlier data (Shuaib et al., 2007). Selfotel, eliprodil, gavestinel, citicoline, magnesium, and a long tail of other candidates followed the same arc. Pharma effectively walked away from the indication around 2010. Any new neuroprotectant inherits the priors that field has earned. A reviewer looking at Semax stroke data starts from a position of trained skepticism toward the entire claim category.
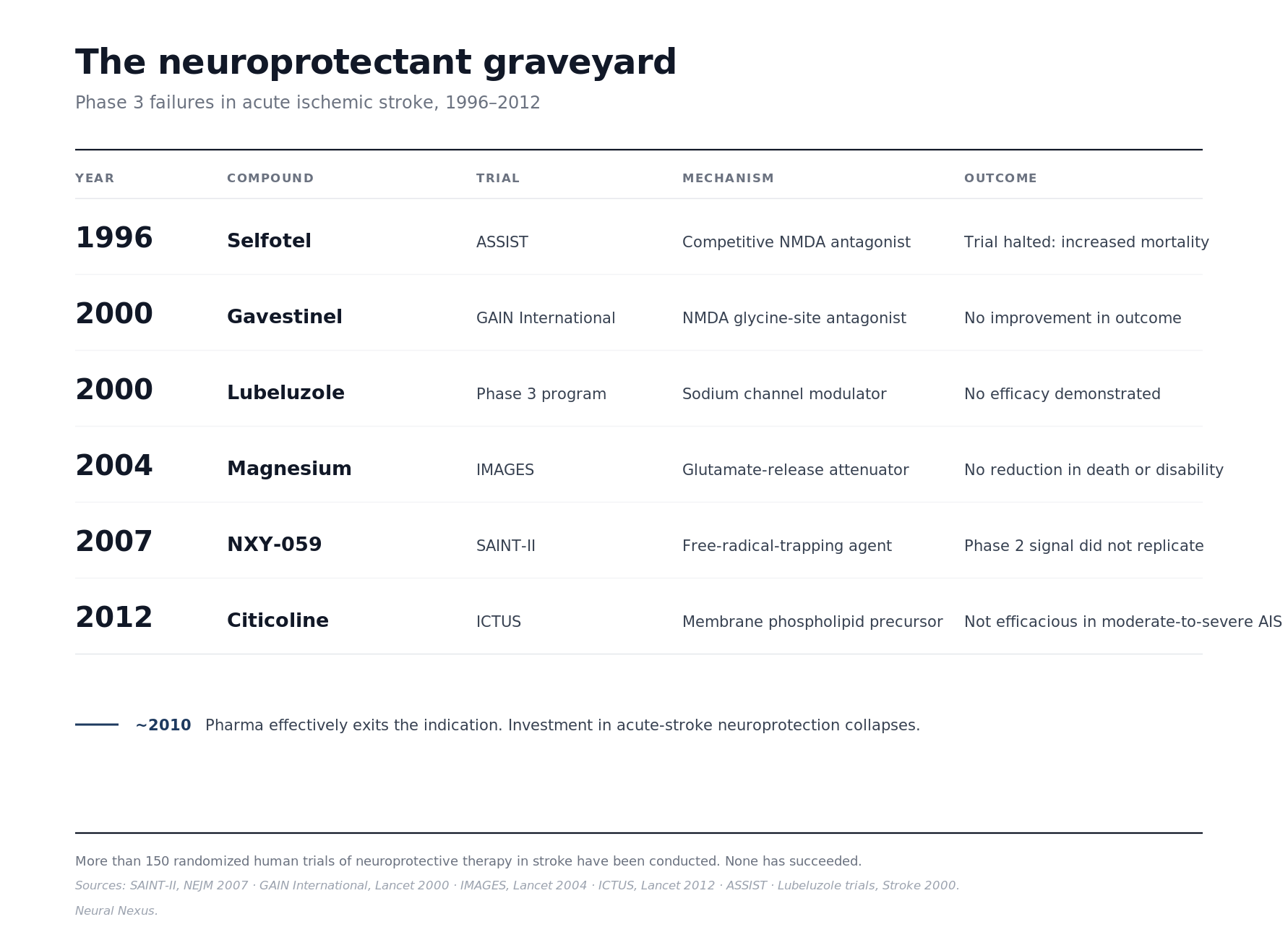
Standard-of-care lock-in. The Russian Semax stroke trials, including the canonical Gusev acute hemispheric stroke study, ran against control arms that received supportive care alone (Gusev et al., 1997). Tissue plasminogen activator was not yet routine. Mechanical thrombectomy did not exist. A modern American trial would have to demonstrate incremental benefit on top of two interventions that already work. The sample sizes and effect-size resolution required to do that are an order of magnitude beyond what the original Russian work approached. A bridging study in 2026 would need larger samples, cleaner endpoints, enough power to detect incremental benefit on top of modern stroke care, and the willingness to accept that the answer may come back negative. This is the one filter where the gate might be tracking real biology rather than infrastructure.
Off-patent and pleiotropic. The familiar problem. Semax is off-patent. There is no exclusivity to recoup development costs. The compound also appears to do several things at once across multiple receptor systems and multiple candidate indications. That confuses a regulatory framework designed to evaluate single compounds against single endpoints in single disease populations. A drug that does several things moderately well does not fit a system designed to prove one thing definitively.
The credibility tax. Semax has been adopted by the biohacker and functional medicine ecosystem and circulates in adjacent wellness culture. The April 2026 reclassification itself is widely characterized in trade press as RFK-Jr.-era HHS direction, which sharpens the political coloring. The same association that drives consumer demand actively repels the institutional actors who would otherwise study it. “Wellness-coded” remains a near-fatal credibility signal in mainstream American medicine. A neurologist who proposes a Semax investigator-initiated trial today is making a career bet that no department chair has any incentive to approve.
The honest counterweight
A serious neurologist looking at the actual published Semax trial data would not say “this is clearly an effective drug being unjustly excluded.” They would say something closer to “this is a plausibly active compound with a thin and methodologically weak evidence base, used at scale in a country whose regulatory standards I do not trust.”
That position is defensible. Russian clinical trial methodology of the 1990s and 2000s was on average, of lower quality than Western trials of the same era. The Semax stroke trials were small. Many were unblinded. Composite endpoints are common in this literature and are gameable. The thirty-year clinical track record reflects breadth of population exposure, not depth of evidence. The mechanism is still officially not fully characterized. The seven filters described above are not a conspiracy. Most of them exist for reasons.
Seven independently defensible filters can still produce a system that systematically cannot process certain categories of compound, regardless of whether those compounds work. That outcome may be acceptable. It is not currently being chosen deliberately.
What the April 2026 reclassification actually does
Read carefully, the FDA’s April 2026 action is small. The agency removed twelve peptide-based bulk drug substances, Semax among them, from Category 2 of the interim 503A bulks list, the bucket reserved for compounds the agency has flagged as potentially raising significant safety concerns. The Pharmacy Compounding Advisory Committee will hold public review meetings in two batches: BPC-157, KPV, MOTs-C, and TB-500 on July 23, 2026; DSIP and Semax on July 24; and the remaining peptides (LL-37, Dihexa, GHK-Cu, PEG-MGF, and Melanotan II) by February 2027 (Orrick, 2026; FDA, 2026).
The agency is clear about what this is and what it is not. Removal from Category 2 does not mean a substance has been evaluated and approved for the 503A bulks list. The peptides are now in regulatory limbo until the PCAC meets and the FDA takes final action. The mechanical reason for the removals is procedural: the original nominators voluntarily withdrew their nominations, and the safety-concern designation could not stand on a withdrawn record.
What the action does not do: it does not create an FDA approval pathway for Semax in stroke. It does not authorize prescription. It does not address any of the seven filters above. It moves a single procedural lever, eligibility for compounding pharmacy review, and does not touch the structural architecture that made Semax a regulatory orphan in the first place. A 55-year-old in Cleveland who has an ischemic stroke on July 25, 2026, the day after the PCAC meeting, will be in exactly the same position as the same patient on July 23.
Why this matters now
The Semax case looks like a curiosity from a particular geopolitical and historical moment. The same filters now apply to a rapidly growing class of compounds the system is structurally unprepared for, and each category trips a recognizable subset of the seven.
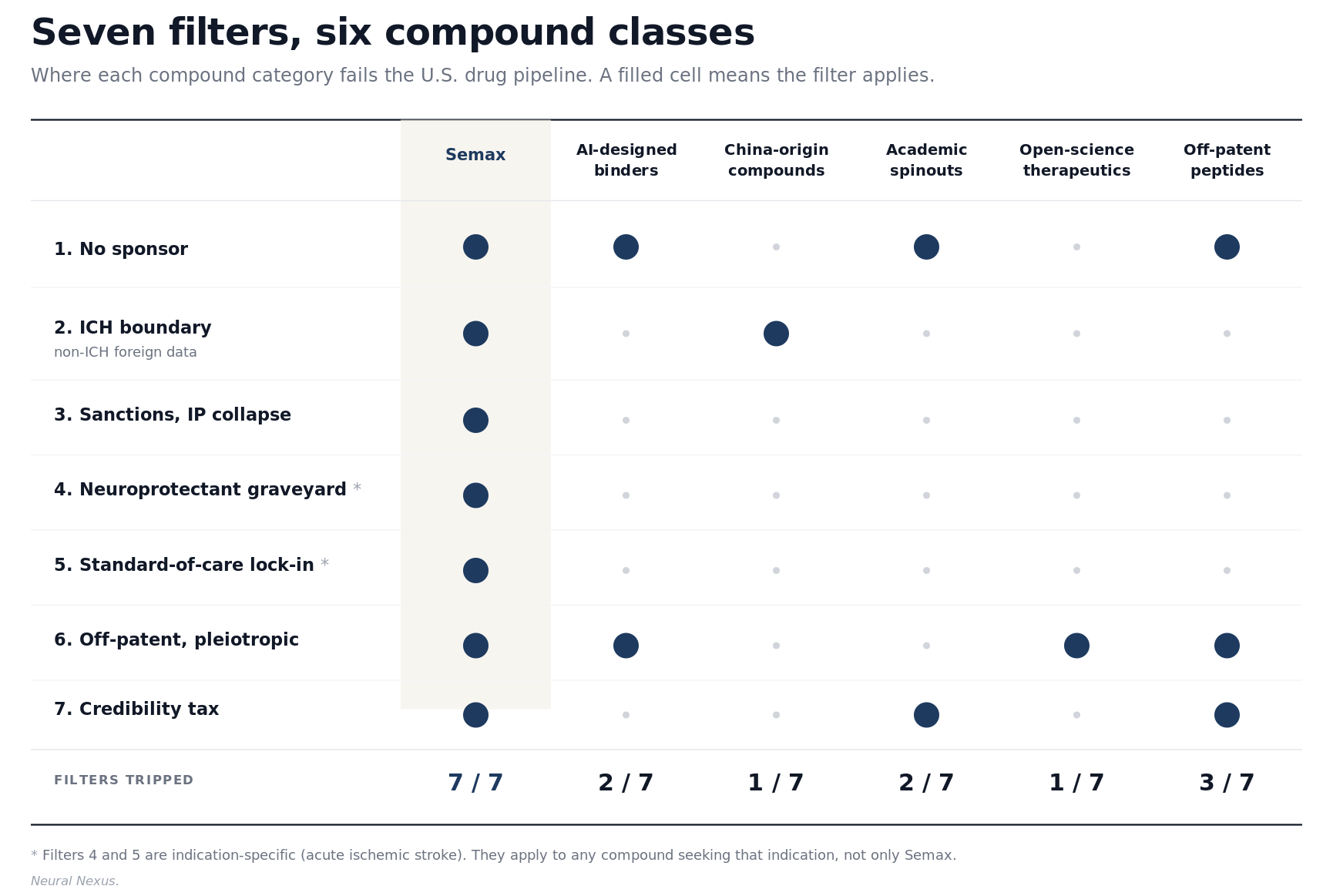
AI-designed protein binders from academic labs trip the no-sponsor filter and the off-patent problem. Compounds from non-ICH jurisdictions, particularly the rising volume now coming out of Chinese clinical infrastructure, trip the foreign-data filter. Academic spinouts whose IP sits inside university tech transfer offices trip the sponsor and credibility filters before they trip anything scientific. Open-science therapeutics released without patent protection by design trip the exclusivity filter as a feature. Off-patent peptide drugs as a class trip the sponsor, exclusivity, and credibility filters simultaneously. None of these categories are exotic. All of them are growing.
The pipeline that was built for sponsor-led, single-indication, ICH-compliant, patent-protected drug development is increasingly mismatched to where useful molecules are now being generated. The April 2026 reclassification is, in this light, the smallest possible regulatory acknowledgment that the off-patent peptide class exists at all. It does not solve the structural problem. It barely registers it.
The conversation about FDA reform tends to focus on speeding approvals or lowering evidentiary bars. Those are real debates. For Semax and the broader category it represents, the binding constraint is upstream. Which evidence the system can metabolize. Which sponsors it requires. Which jurisdictions it recognizes. None of these constraints were chosen on first principles. They accumulated.
The reform conversation that matters is the one about that accumulation. The agency needs an evidence framework for compounds that arrive without a sponsor to package the data. It needs an audit pathway for non-ICH clinical evidence that does not require years of bridging studies. It needs a regulatory route for compounds whose value lies in pleiotropy rather than in a single primary endpoint. It needs an answer for what drug development looks like when molecules are coming from algorithms and university labs faster than pharma can license them.
None of these questions have answers right now. None of them are getting asked at the level of the agency or the legislature. The drug a Cleveland neurologist cannot prescribe to a stroke patient today is not the most important fact in this story. The drug a Cleveland neurologist will not be able to prescribe five years from now, when that drug exists and was designed by an algorithm in an academic lab and has trial data from Beijing, is the more important fact.
The U.S. system has structural blindness to certain categories of evidence and certain origins of molecules. The cost of that blindness is borne by patients who never learn what they were never offered. The science will not settle itself. The system has to decide what it wants to be able to know.
These newsletters take significant effort to put together and are totally for the reader’s benefit. If you find these explorations valuable, there are multiple ways to show your support:
Engage: Like or comment on posts to join the conversation.
Subscribe: Never miss an update by subscribing to the Substack.
Share: Help spread the word by sharing posts with friends directly or on social media.
References
FDA. Certain Bulk Drug Substances for Use in Compounding That May Present Significant Safety Risks (interim 503A list). Updated April 22, 2026.
Gusev EI, Skvortsova VI, Miasoedov NF, Nezavibat’ko VN, Zhuravleva EIu, Vanichkin AV. Effectiveness of semax in acute period of hemispheric ischemic stroke (a clinical and electrophysiological study). Zh Nevrol Psikhiatr Im S S Korsakova. 1997;97(6):26-34.
Orrick, Herrington & Sutcliffe LLP. FDA Announces Removal of 12 Peptides from Category 2 and Schedules PCAC Meetings to Consider Adding Peptides to 503A Bulk Drug Substances List. April 2026.
Potaman VN, Alfeeva LY, Kamensky AA, Levitzkaya NG, Nezavibatko VN. N-terminal degradation of ACTH(4-10) and its synthetic analog semax by the rat blood enzymes. Biochemical and Biophysical Research Communications. 1991;176:741-746.
Shuaib A, Lees KR, Lyden P, et al; SAINT II Trial Investigators. NXY-059 for the treatment of acute ischemic stroke. New England Journal of Medicine. 2007;357:562-571. doi:10.1056/NEJMoa070240