
BioWire Byte 024 - A Liver Enzyme Reverses Brain Aging. Exercise Turns It On.
Paper traces a complete molecular chain from the liver to the blood-brain barrier to cognition

Physically active adults develop dementia at lower rates, perform better on cognitive tests, and show less hippocampal atrophy on MRI. The epidemiology has been clear for years. What hasn’t been clear is the mechanism. Researchers have proposed BDNF, irisin, and a half-dozen other mediators. None of them traced a complete chain from a peripheral organ to the brain and back to a cognitive outcome. A study published in Cell in February 2026 does exactly that, and the organ in question is the liver (Bieri, Pratt et al., Cell, 2026).
First, if you enjoy these posts, consider subscribing and becoming a part of our growing community!
The connection starts with an enzyme called GPLD1, a phospholipase secreted by the liver whose circulating levels rise with exercise. Saul Villeda’s lab at UCSF identified it in 2020 by transferring plasma from exercised mice into sedentary aged mice, then running proteomics on the blood to find what was doing the work (Horowitz et al., Science, 2020). Overexpressing GPLD1 in the liver of aged mice recapitulated the cognitive benefits of exercise plasma transfer. But GPLD1 does not cross the blood-brain barrier. Whatever it was doing, it was doing it from the outside.
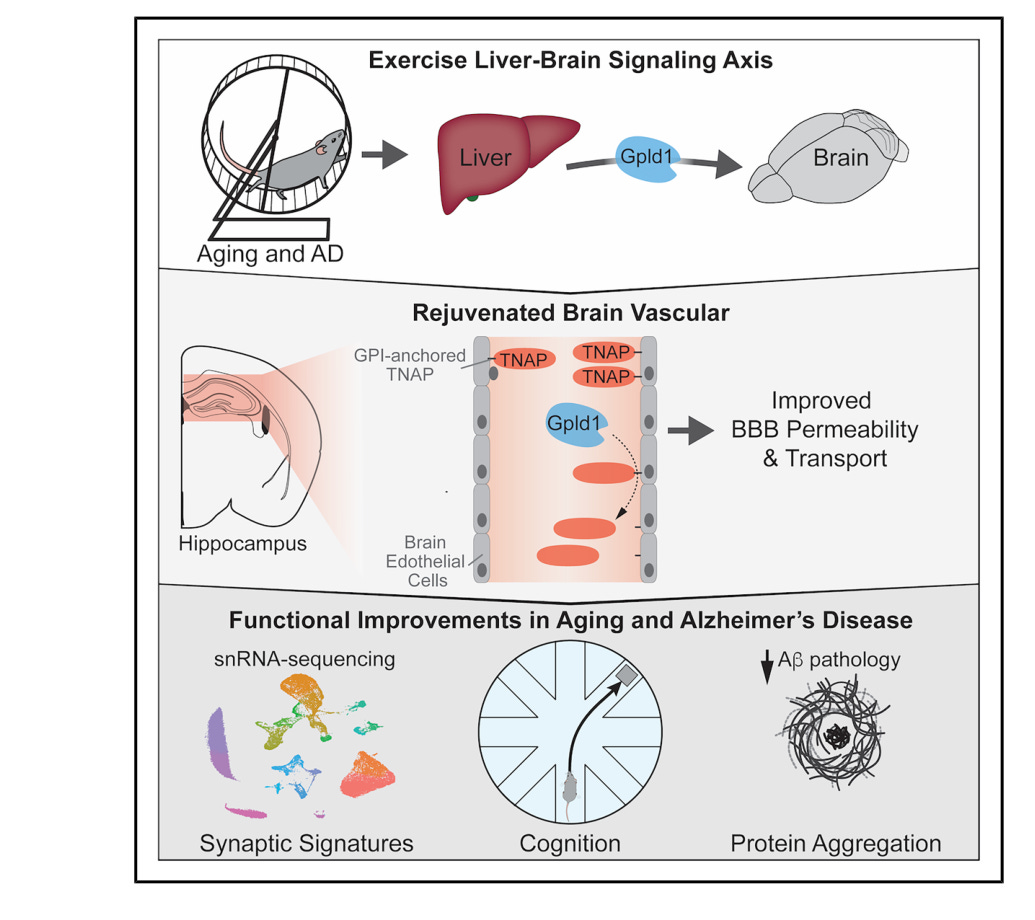
The new paper identifies the target. TNAP, tissue-nonspecific alkaline phosphatase, is a protein anchored to the surface of brain endothelial cells, the cells that form the blood-brain barrier (BBB). As the brain ages, TNAP accumulates on these cells. GPLD1 cleaves it off, physically stripping TNAP from the membrane. Circulating TNAP levels rise in the blood after treatment, confirming the cleavage is happening in real time.
The consequences are immediate. Aged mice show extensive tracer leakage from hippocampal blood vessels. A barrier in disrepair. GPLD1 treatment reduced that leakage across the hippocampus and reversed a large fraction of age-related gene expression changes in brain endothelial cells, pushing them back toward a youthful profile. TNAP inhibition alone reproduced the majority of those effects. Two different interventions targeting the same enzyme, same transcriptomic direction.
To prove TNAP itself drives the decline, the group engineered young, healthy mice to overexpress it on brain endothelial cells. Those animals developed barrier leakage and cognitive impairment at four to five months old. TNAP accumulation alone was sufficient to age the brain’s vasculature. Running the experiment in reverse, CRISPR knockout of TNAP in aged mice improved cognition. So did SBI-425, an oral TNAP inhibitor that never crosses the blood-brain barrier. The rescue works from the periphery.
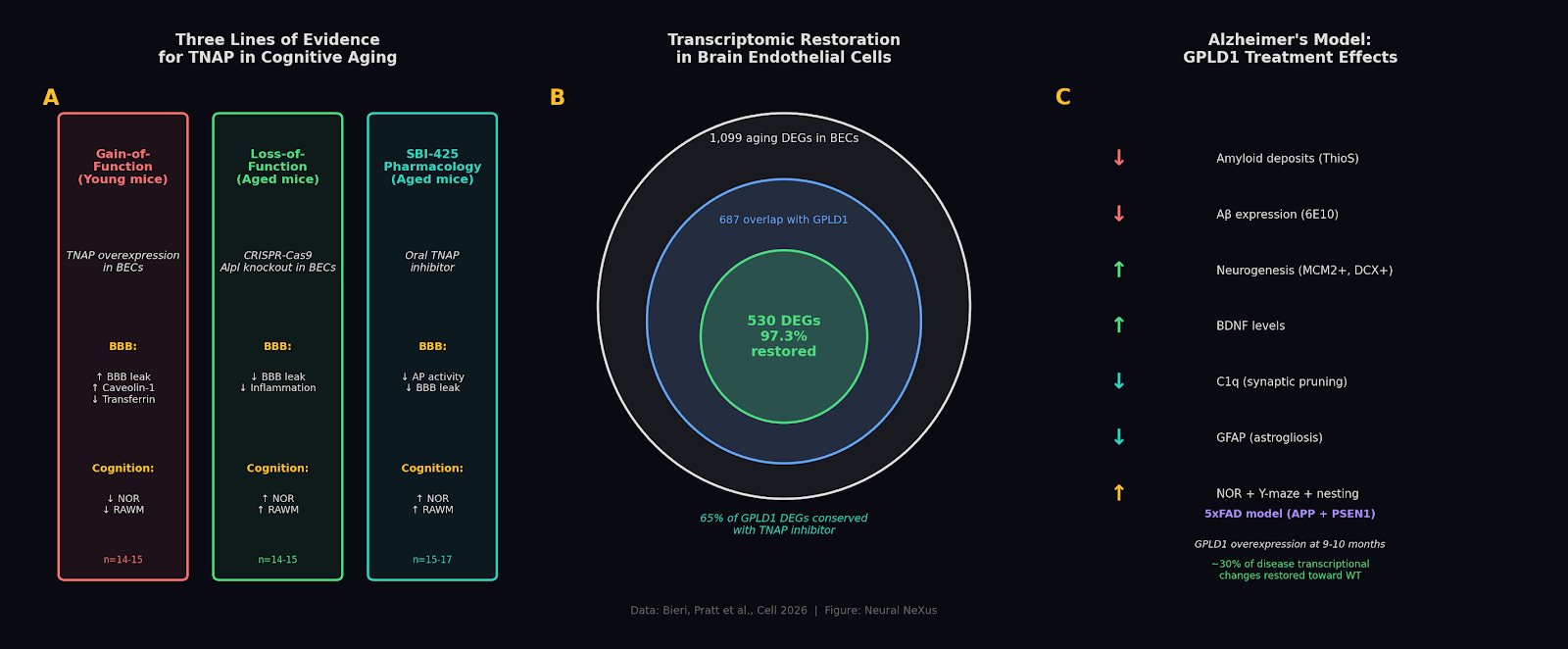
The group then asked whether this holds in disease, not just aging. In a mouse model of Alzheimer’s disease, GPLD1 treatment reduced amyloid deposits, dampened neuroinflammation, and improved cognition across multiple behavioral tests. SBI-425 replicated most of those benefits but missed one, Y-maze working memory, which GPLD1 rescued. That gap matters because GPLD1 cleaves over 100 proteins beyond TNAP, and its full cognitive reach appears broader than any single target. For drug development, that’s a feature. TNAP inhibition gives you a clean peripheral target for barrier repair, while the biology leaves room for combination strategies that capture more of what exercise actually does.
The medical research field has spent decades searching for a drug that meaningfully rescues cognitive decline. Most candidates fail because the brain is hard to reach. SBI-425 sidesteps that problem entirely. It’s oral, it never crosses the blood-brain barrier, and it has already shown cognitive benefits in mice. That profile is cleaner than most CNS drug candidates, though peripheral TNAP plays roles in other tissues and those liabilities would need evaluation. (Disclosure: Villeda is co-founder of Ceiba Bio, Inc.; the work is covered by patent PCT/US2020/016549.)
Early human data hints in the same direction. Western blots of human cortical tissue showed TNAP protein levels elevated in older adults and Alzheimer’s patients compared with healthy young controls. The broader implication is uncomfortable for a field that has spent decades treating the brain as the protagonist of its own decline. Amyloid is made in neurons. Tau tangles spread through neural circuits. Inflammation is driven by resident microglia. All of that is framed as intrinsic failure. This paper suggests the brain may be more dependent on peripheral vascular support than anyone assumed. A liver enzyme, elevated by running, can shift hundreds of age-related endothelial gene expression changes back toward youth and improve cognition in aged and Alzheimer’s mice. If the barrier is the bottleneck, how much of what we call brain aging is actually vascular aging that the brain merely suffers?
No one has answered that in humans yet. But if SBI-425 achieves in humans even a fraction of what it does in mice, the field will need to reckon with a possibility it has largely set aside: that the best way to treat a failing brain might be to fix the pipes.
These newsletters take significant effort to put together and are totally for the reader’s benefit. If you find these explorations valuable, there are multiple ways to show your support:
Engage: Like or comment on posts to join the conversation.
Subscribe: Never miss an update by subscribing to the Substack.
Share: Help spread the word by sharing posts with friends directly or on social media.
References
Bieri G, Pratt K, Fuseya Y, et al. Liver exerkine reverses aging- and Alzheimer’s-related memory loss via vasculature. Cell. 2026;189(4). doi: 10.1016/j.cell.2026.01.024
Horowitz AM, Fan X, Bieri G, et al. Blood factors transfer beneficial effects of exercise on neurogenesis and cognition to the aged brain. Science. 2020;369(6500):167-173. doi: 10.1126/science.aaw2622
Another goddamned reason to exercise. Will it never end?